Sequana_pipetools documentation¶

- Overview:
A set of tools to help building or using Sequana pipelines
- Status:
Production
- Issues:
Please fill a report on github
- Citation:
Cokelaer et al, (2017), ‘Sequana’: a Set of Snakemake NGS pipelines, Journal of Open Source Software, 2(16), 352, JOSS DOI doi:10.21105/joss.00352
🔧 Installation¶
from pypi website:
pip install sequana_pipetools
No dependencies for this package except Python packages. In practice, this package has no interest if not used within a Sequana pipeline. It is installed automatically when you install a Sequana pipelines. For example:
pip install sequana_rnaseq
pip install sequana_fastqc
See Sequana for a list of pipelines ready for production.
🎯 Targetted audience¶
This package is intended for Sequana developers seeking to integrate Snakemake pipelines into the Sequana project. Please refer below for more information. Additionally, note that as a developer, you can generate the reference documentation using Sphinx:
git clone https://github.com/sequana/sequana_pipetools
cd html
make html
browse build/html/index.html
❓ What is sequana_pipetools ?¶
sequana_pipetools is a collection of tools designed to facilitate the management of Sequana pipelines, which includes next-generation sequencing (NGS) pipelines like RNA-seq, variant calling, ChIP-seq, and others.
The aim of this package is to streamline the deployment of Sequana pipelines by creating a pure Python library that includes commonly used tools for various pipelines.
Previously, the Sequana framework incorporated all bioinformatics, Snakemake rules, pipelines, and pipeline management tools into a single library (Sequana) as illustrated in Fig 1 below.
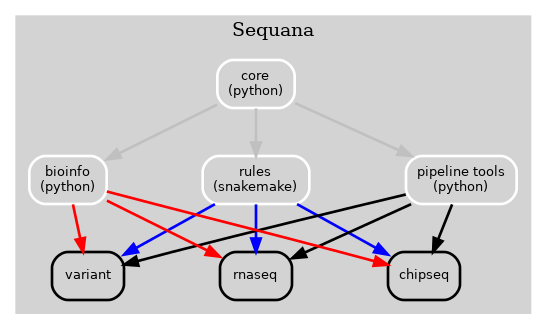
Figure 1 Old Sequana framework will all pipelines and Sequana library in the same place including pipetools (this library).¶
Despite maintaining an 80% test coverage, whenever changes were introduced to the Sequana library, a comprehensive examination of the entire library was imperative. The complexity escalated further when incorporating new pipelines or dependencies. To address this challenge, we initially designed all pipelines to operate independently, as depicted in Fig. 2. This approach allowed modifications to pipelines without necessitating updates to Sequana and vice versa, resulting in a significant improvement.
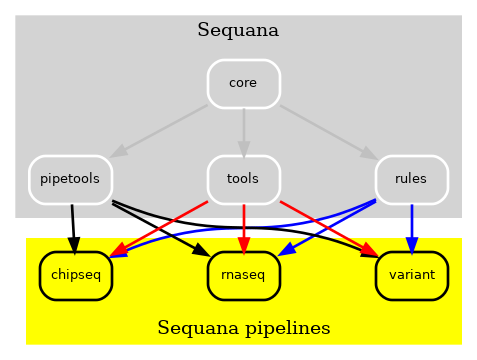
Figure 2 v0.8 of Sequana moved the Snakemake pipelines in independent repositories. A cookie cutter ease the creation of such pipelines¶
Nevertheless, certain tools, including those utilized for user interface and input data sanity checks, were essential for all pipelines, as illustrated by the pipetools box in the figure. With the continuous addition of new pipelines each month, our goal was to enhance the modularity of both the pipelines and Sequana. As a result, we developed a pure Python library named sequana_pipetools, depicted in Fig. 3, to further empower the autonomy of the pipelines.
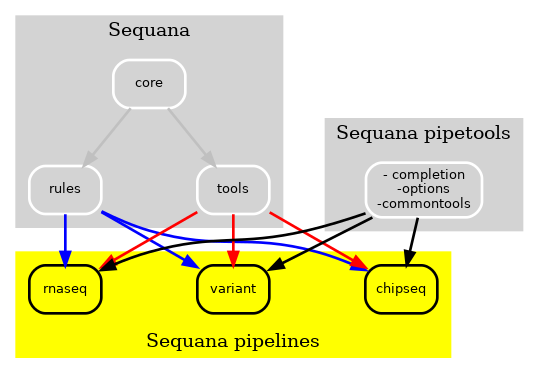
Figure 3 New Sequana framework. The new Sequana framework comprises the core library and bioinformatics tools, which are now separate from the pipelines. Moreover, the sequana_pipetools library provides essential tools for the creation and management of all pipelines, including a shared parser for options¶
As a final step, we separated the rules originally available in Sequana to create an independent package featuring a collection of Snakemake wrappers. These wrappers can be accessed at https://github.com/sequana/sequana-wrappers and offer the added benefit of being rigorously tested through continuous integration.
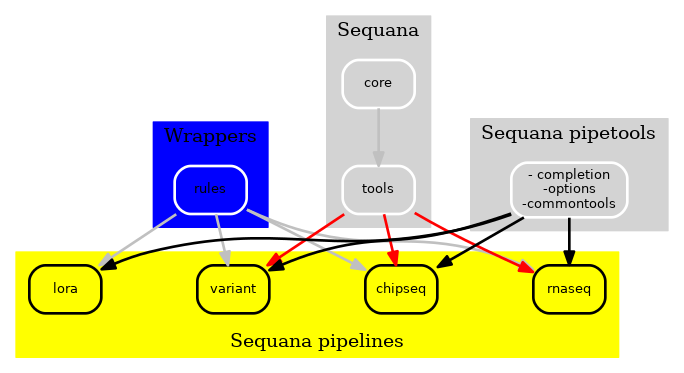
Figure 3 New Sequana framework 2021. The library itself with the core, the bioinformatics tools is now fully independent of the pipelines.¶
Quick tour of the standalone¶
The sequana_pipetools package provide a standalone called sequana_pipetools. Here is a snapshot of the user interface:
There are several applications. The main one is used to initiate pipeline skeleton and tree structure automatically using:
sequana_pipetools --init-new-pipeline
See below for more details.
Then, we provide some utilities. For instance, for Linux users, under bash shell, you can setup the completion of a sequana pipeline command line arguments:
sequana_pipetools --completion fastqc
The second is used to introspect slurm files to get a summary of the SLURM log files:
sequana_pipetools --slurm-diag
It searches for files with pattern slurm in the current directory and slurm files in the ./logs directory. This is used within th pipeline but can be used manually as well and is useful to get a quick summary of common errors found in slurm files.
The following command provides statistics about Sequana pipelines installed on your system (number of rules, wrappers used):
sequana_pipetools --stats
And for developpers, a quick creation of schema file given a config file (experimental, developers would still need to edit the schema but it does 90% of the job):
sequana_pipetools --config-to-schema config.yaml > schema.yaml
You can also convert the dot file into a nice PNG file using:
sequana_pipetools --dot2png dag.dot
To diagnose pipeline errors using an LLM (requires a Mistral or OpenAI API key):
sequana_pipetools --diagnose
To monitor a running pipeline with a rich progress display (used internally by pipelines launched with --monitor):
sequana_pipetools --monitor
For Sequana developers¶
The library is intended to help Sequana developers to design their pipelines. See the Sequana organization repository for examples. In addition to the standalone shown above, sequana_pipetools main goal is to provide utilities to help Sequana developers.
First, let us create a pipeline
Update the main script¶
Go to sequana_pipelines/NAME and look at the main.py script.
We currently provide a set of Options classes that should be used to design the API of your pipelines. For example, the sequana_pipetools.options.SlurmOptions can be used as follows inside a standard Python module (the last two lines is where the magic happens):
import rich_click as click
from sequana_pipetools.options import *
from sequana_pipetools import SequanaManager
NAME = "fastqc"
help = init_click(NAME, groups={
"Pipeline Specific": [
"--method", "--skip-multiqc"],
}
)
@click.command(context_settings=help)
@include_options_from(ClickSnakemakeOptions, working_directory=NAME)
@include_options_from(ClickSlurmOptions)
@include_options_from(ClickInputOptions, add_input_readtag=False)
@include_options_from(ClickGeneralOptions)
@click.option("--method", default="fastqc", type=click.Choice(["fastqc", "falco"]), help="your msg")
def main(**options):
# the real stuff is here
manager = SequanaManager(options, NAME)
manager.setup()
# just two aliases
options = manager.options
cfg = manager.config.config
# fills input_data, input_directory, input_readtag
manager.fill_data_options()
# fill specific options.
# create a function for a given option (here --method)
def fill_method():
# any extra sanity checks
cfg["method"] = options["method"]
if options["from-project"]:
# in --from-project, we fill the method is --method is provided only (since already pre-filled)
if "--method" in sys.argv
fill_method()
else:
# in normal, we always want to fill the user-provided option
fill_method()
# finalise the command and save it; copy the snakemake. update the config
# file and save it.
manager.teardown()
if __name__ == "__main__":
main()
Developers should look at e.g. module sequana_pipetools.options for the API reference and one of the official sequana pipeline (e.g., https://github.com/sequana/sequana_variant_calling) to get help from examples.
The Options classes provided can be used and combined to design pipelines.
How to use sequana pipetools within your Pipeline¶
For FastQ files (paired ot not), The config file should look like:
sequana_wrappers: "v0.15.1"
input_directory: "."
input_readtag: "_R[12]_"
input_pattern: "*fastq.gz"
apptainers:
fastqc: "https://zenodo.org/record/7923780/files/fastqc_0.12.1.img"
section1:
key1: value1
key2: value2
And your pipeline could make use of this as follows:
configfile: "config.yaml"
from sequana_pipetools import PipelineManager
manager = PipelineManager("fastqc", config)
# you can then figure out wheter it is paired or not:
manager.paired
# get the wrapper version to be used within a rule:
manager.wrappers
# the raw data (with a wild card) for the first rule
manager.getrawdata()
# add a Makefile to clean things at the end
manager.teardown()
Setting up and Running Sequana pipelines¶
When you execute a sequana pipeline, e.g.:
sequana_fastqc --input-directory data
a working directory is created (with the name of the pipeline; here fastqc). Moreover, the working directory contains a shell script that will hide the snakemake command. This snakemake command with make use of the sequana wrappers and will use the official sequana github repository by default (https://github.com/sequana/sequana-wrappers). This may be overwritten. For instance, you may use a local clone. To do so, you will need to create an environment variable:
export SEQUANA_WRAPPERS="git+file:///home/user/github/sequana-wrappers"
If you decide to use singularity/apptainer, one common error on a cluster is that non-standard paths are not found. You can bind them using the -B option but a more general set up is to create this environment variable:
export SINGULARITY_BINDPATH="/path_to_bind"
for Apptainer setup
export APPTAINER_BINDPATH="/path_to_bind"
What is Sequana ?¶
Sequana is a versatile tool that provides
A Python library dedicated to NGS analysis (e.g., tools to visualise standard NGS formats).
A set of Pipelines dedicated to NGS in the form of Snakefiles (Makefile-like with Python syntax based on snakemake framework) with more common wrappers.
Standalone applications such as sequana_coverage and sequana_taxonomy.
See the sequana home page for details.
To join the project, please let us know on github.
Changelog :memo:¶
Version |
Description |
|---|---|
1.5.7 |
|
1.5.6 |
|
1.5.5 |
|
1.5.3 |
|
1.5.2 |
|
1.5.1 |
|
1.5.0 |
|
1.4.0 |
|
1.3.1 |
|
1.3.0 |
|
1.2.2 |
|
1.2.1 |
|
1.2.0 |
|
1.1.1 |
|
1.1.0 |
|
1.0.X |
|
1.0.0 |
|
0.17.X |
|
0.16.9 |
|
0.16.8 |
|
0.16.7 |
|
0.16.6 |
|
0.16.5 |
|
0.16.4 |
|
0.16.3 |
|
0.16.2 |
|
0.16.1 |
|
0.16.0 |
|
0.15.X |
|
0.14.X |
|
0.13.0 |
|
0.12.X |
|
0.11.X |
|
0.10.X |
|
0.9.X |
|
0.9.0 |
|
0.8.X |
|
0.7.X |
|
0.6.X |
|
0.5.X |
|
0.4.X |
|
0.4.0 |
|
0.3.X |
|
0.2.X |
|
0.1.X |
|
- left_speech_bubble:
Contacts <a name="contacts"></a>
- question:
Feel free to [open an issue](https://github.com/sequana/sequana_pipetools/issues)
- References
- Options
- Sequana Manager
- FileFactory and FastQFactory
- Pipeline manager
PipelineManagerPipelineManagerBasePipelineManagerBase.clean_multiqc()PipelineManagerBase.error()PipelineManagerBase.get_html_summary()PipelineManagerBase.get_run()PipelineManagerBase.get_shell()PipelineManagerBase.getmetadata()PipelineManagerBase.getrawdata()PipelineManagerBase.onerror()PipelineManagerBase.onsuccess()PipelineManagerBase.setup()PipelineManagerBase.snakefilePipelineManagerBase.teardown()
PipelineManagerDirectoryget_run()get_shell()